Spletna revija za znanstvenike, strokovnjake
in nevroznanstvene navdušence
Naslovnica Članki Intervjuji Mnenja Zdravje Korenine eSinapsa Številke ![]()
Konformacijske bolezni
letnik 2011, številka 3
uvodnik
članki
Mara Bresjanac
Martina Starc
Rok Berlot
Varnost uporabe generičnih protiepileptičnih zdravil
Mojca Kržan, Matevž Kržan
Možgani, računalniki - nekaj vmes
Miha Pelko
aktualno
Stroški bolezni možganov v Evropi v 2010
Jure Bon, Zvezdan Pirtošek, Blaž Koritnik in Mara Bresjanac
Poročilo s Foruma mladih nevroznanstvenikov v Ljubljani
Simon Brezovar
mnenje
kolofon
letnik 2011, številka 3
Ste se morda vprašali “Kaj se je zgodilo z boleznijo norih krav?” Po vznemirjenju, ki so ga ob koncu prejšnjega stoletja sprožili prioni, ko so po bolezni pri kravah povzročili tudi epidemijo t.i. variante Creutzfeldt Jakobove bolezni pri ljudeh, o teh boleznih v medijih skorajda ni več slišati. A ne zato, ker bi ne bile aktualne. Raziskave so razkrile, da so nekatere pogoste bolezni, kot je denimo Alzheimerjeva bolezen, presenetljivo podobne prionskim po mehanizmih nastanka, kar odpira nove diagnostične in terapevtske možnosti.
 Medicina razvršča bolezni po različnih kriterijih, od tega, kateri organ(ski sistem) je primarno prizadet, do tega, katera stroka je primarno udeležena v zdravljenju ali lajšanju simptomov bolezni. Ko gre za motnje v delovanju možganov, imamo tako opravka z nevrološkimi na eni in psihiatričnimi stanji na drugi strani. S poglabljanjem razumevanja patofiziologije se tovrstne razmejitve postopno umikajo. Namesto njih postajajo kriterij za nove razvrstitve in temelj za razvoj diagnostičnih in terapevtskih pristopov patogenetski mehanizmi, ki so sorodni sicer navidez zelo raznolikim boleznim.
Medicina razvršča bolezni po različnih kriterijih, od tega, kateri organ(ski sistem) je primarno prizadet, do tega, katera stroka je primarno udeležena v zdravljenju ali lajšanju simptomov bolezni. Ko gre za motnje v delovanju možganov, imamo tako opravka z nevrološkimi na eni in psihiatričnimi stanji na drugi strani. S poglabljanjem razumevanja patofiziologije se tovrstne razmejitve postopno umikajo. Namesto njih postajajo kriterij za nove razvrstitve in temelj za razvoj diagnostičnih in terapevtskih pristopov patogenetski mehanizmi, ki so sorodni sicer navidez zelo raznolikim boleznim.
Primer je skupina konformacijskih bolezni (preglednica 1). Tako sta jih poimenovala1, ki sta jih opredelila kot stanja, do katerih pride, ko neka beljakovina iz svoje nativne, funkcionalne oblike spremeni konformacijo tako, da njene molekule zavzamejo obliko (konformacijo) beta-nagubanega lista in se pričnejo zlagati ena na drugo. Tako nastanejo strukturno zelo urejeni, netopni, vlaknati polimeri, ki imajo pomembne skupne lastnosti ne glede na to, iz katere beljakovine so nastali. Generično ime za te polimere je amiloid, kar izpostavlja njihove skupne ultrastrukturne, biokemijske in histokemijske lastnosti: tvorijo jih nerazvejana vlakna premera 7,5 do 10 nm, amiloidne odlage so kongofilne (obarvajo se z barvilom Kongo rdeče) in v polarizacijskem mikroskopu kažejo zeleno dvolomnost 2. Odlage amiloida so značilnost vseh konformacijskih bolezni, ki pa se razlikujeje po tipu odlag (npr. pri Alzheimerjevi bolezni najdemo beta amiloid v plakih med celicami, beljakovina tau pa se odlaga v obliki pentelj znotraj celic), po tem, v katerih tkivih in organih se odlaga amiloid in po klinični sliki bolezni. Za ilustracijo izpostavimo le nekatere primere raznolikih bolezni, ki jim je skupna konformacijska patologija: sladkorna bolezen tip 2, cistična fibroza, dedni panlobularni emfizem, Alzheimerjeva in Parkinsonova bolezen.
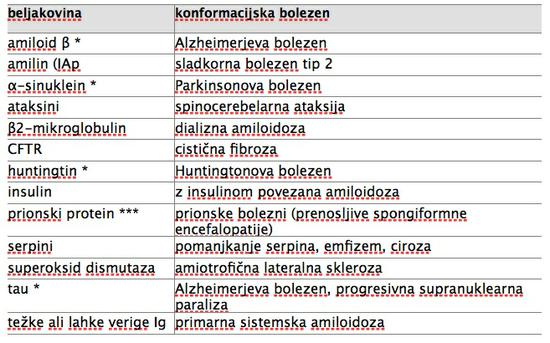
Preglednica 1: Primeri konformacijskih bolezni in beljakovin, ki se pri the boleznih odlagajo. Ena zvezdica * označuje beljakovine, ki so prenosljive na molekularni ravni (t.i. prionoidi), tri zvezdice *** pa označujejo prion, primer bolezensko spremenjene beljakovine, ki je potrjeno kužna po vnosu v organizem z uživanjem hrane s prioni, s presaditvijo organov, invazivnimi posegi v živčevje, s transfuzijo krvi ter eksperimentalno celo po vdihavanju aerosolov s prioni.6
Konformacijska sprememba, ki je podlaga bolezenskemu združevanju in odlaganju beljakovin, naj bi bila prisotna tudi v normalnih okoliščinah kot prehodno in reverzibilno stanje proteinske homeostaze – proteostaze3 (slika 1). Verjetnost nastanka bolezni se poveča, kadar se ravnovesje med nastajanjem in razgradnjo beljakovine prevesi v smer kopičenja. Pri tem lahko konformacijska sprememba povzroči izgubo funkcije nativne beljakovine (tak primer je izguba funkcije transmembranske beljakovine CFTR pri cistični fibrozi). Po drugi strani bolezensko zvita beljakovina lahko pridobi novo, toksično funkcijo (npr. amiloid beta pri Alzheimerjevi bolezni), ali pa v nastanku tkivne okvare in klinične bolezni sodelujeta izguba normalne in pridobitev toksične funkcije.
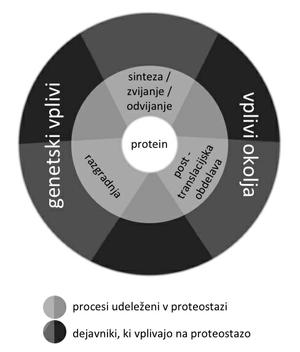
Slika 1: Homeostaza proteinov – proteostaza – so procesi sinteze, post-translacijske obdelave in razgradnje beljakovin, ki vzdržujejo njihovo normalno koncentracijo in delovanje. Genetski dejavniki ter okolje, ki spreminja razmere v organizmu (npr. stres, kalorični vnos …), vplivajo na proteostazo neposredno in posredno.
Okoliščine, ki rušijo proteostazo, so lahko prirojene, kot denimo genske mutacije ter polimorfizmi, ki vplivajo na stabilnost in zvijanje beljakovin, ali pa gre za različne vplive okolja na organizem. Poleg vnetnih procesov, oksidativnega stresa in presnovnih dejavnikov, ki lahko olajšajo in pospešijo razvoj konformacijskih bolezni, lahko sem štejemo še njihovo prenosljivost: bolezensko preobrazbo lahko sproži vnos patološko spremenjene beljakovine v predhodno zdrav organizem. Ker je ves patogeni potencial vsebovan le v strukturnih značilnostih, t.j. v konformaciji beljakovine, pri teh boleznih niso izpolnjeni temeljni Kochovi kriteriji, da bi jih lahko opredelili za “nalezljive”. Prehod nativne beljakovine v bolezensko konformacijo po stiku z patološko beljakovino je v nekaterih primerih dokumentirana le na molekularni ravni (npr. pri amiloidu beta4 in alfa-sinukleinu5). Za razliko od prionov, ki lahko povzročijo bolezen po katerikoli poti vstopanja v organizem6, se zato za amiloid beta, alfa-sinuklein, tau in druge amiloidogene beljakovine, ki so prenašalci konformacijske patologije le ob neposrednem vnosu v dovzetni organ ali tkivo, uveljavlja ime prionoidi7.
Namesto, da bi bile prionske bolezni pozabljene, so torej prav raziskave prionov prispevale k boljšemu razumevanju patofiziologije cele vrste konformacijskih bolezni, odprl pa se je tudi boljši vpogled v primere v biologiji, ki kažejo, da so konformacijske spremembe z nastankom amiloida v določenih razmerah del normalne fiziologije. Primer za fiziološke amiloide je npr. oblikovanje dologoročnih spominskih sledi v aktivnih sinapsah8 prek trajne konformacijske spremembe ustreznih sinaptičnih beljakovin ali denimo skladiščenje hormonov9.
Čeprav konformacijskih bolezni danes še ne znamo zdraviti, boljše razumevanje mehanizma njihovega nastanka odpira pomembne nove smeri za njihovo zgodnjo prepoznavo in razvoj zdravljenja.
-
___
-
Carrell RW, Lomas DA. Conformational disease. Lancet 1997; 350: 134 - 8. ↩
-
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Eng J Med 2003;349:583-96. ↩
-
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009; 78:959-91. ↩
-
Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci 2000; 20, 3606–11. ↩
-
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synu- clein. Proc Natl Acad Sci USA 2009; 106, 13010–5. ↩
-
Haybaeck J, Heikenwalder M, Klevenz B, Schwarz P, Margalith I, Bridel C, Mertz K, Zirdum E, Petsch B, Fuchs TJ, Stitz L, Aguzzi A. Aerosols Transmit Prions to Immunocompetent and Immunodeficient Mice. PLoS Pathog 7(1): e1001257. doi:10.1371 journal.ppat.1001257. ↩
-
Aguzzi A. Cell biology: Beyond the prion principle. Nature 2009; 459, 924–925. ↩
-
Si K, Lindquist S, Kandel ER. A Neuronal Isoform of the Aplysia CPEB Has Prion-Like Properties. Cell 2003; 115: 879-9. ↩
-
Greenwald J, Riek R. Biology of Amyloid: Structure, Function, and Regulation. Structure 2010; 18: 1244-60. ↩
prof. dr. Mara Bresjanac
Laboratorij za regeneracijo in plastičnost živčevja
Inštitut za patološko fiziologijo, Medicinska fakulteta Univerze v Ljubljani